On April 4, 1951, a 22-year-old military inductee stumbled into a U.S. naval hospital in Philadelphia. Doubled over with acute back and abdominal pain, he could barely walk. As nurses tried to secure him to a gurney, his nose began to bleed profusely; a flood of red spilled over his clothes. After several hours doctors managed to stabilize him, but they couldn’t figure out what had caused the bleeding. Some suspected he had been poisoned. But with what? And by whom?

An undated photo of University of Wisconsin biochemist Karl Paul Link, whose lab first isolated the powerful blood thinner and rat poison warfarin.
When the young recruit was finally lucid, doctors pried the story out of him. Depressed by his future prospects and desperate to avoid deployment at any cost, he had taken a small amount of rat poison. The next morning, to his shock, he woke up alive and well. Doggedly, he swallowed the poison again that night and for the next four nights, to no avail. Then on April 2 a sudden sharp pain set in along his torso, and blood began to spurt from his nose. He thought he was finally dying. But two days later, as the pain became overwhelming, he realized (to his consternation) he was still very much alive.
The recruit was despondent as he told his tale, but his doctors were intrigued. They published his curious case in medical journals, and EJH (as he was dubbed) became part of medical history. How had this man survived 500 milligrams of rat poison that would have killed a rat many times over and probably most other humans? This question would occupy medical researchers into the 21st century.
EJH’s poison of choice was a commercial rat bait known as warfarin, sold under the brand name d-CON. Warfarin had made its market debut in the early 1950s and almost instantly became a familiar sight in homes, businesses, schools, and places of worship across the United States. EJH reasoned if it could kill rats, it could kill humans too. What he couldn’t imagine is that his brush with death would help save countless lives.
Warfarin was discovered by researchers attempting to solve yet another medical mystery, this one affecting cattle. In the early 1920s a strange and bloody illness slowly spread through cattle farms across Canada and the United States. Farmers watched in horror as their otherwise healthy cows suddenly became sick—often after routine procedures, such as dehorning—collapsing in pools of their own blood. The disease wiped out entire herds, destroyed farms and rural livelihoods, and baffled veterinarians for a decade.
Then, during a blustery Wisconsin snowstorm in February 1933, a frustrated farmer from Deer Park named Ed Carlson braved the 200-mile journey to the state capital, Madison. In the back of his truck he had stashed a dead cow, a bucket of its unclotted blood, and a hundred pounds of moldy hay. A veterinarian had recommended that the farmer consult experts at the University of Wisconsin’s Agricultural Experiment Station, and chance brought him late that Saturday afternoon to the lab of biochemist Karl Paul Link and his students, who were working after hours. Carlson demanded to know why his prized bulls and cows were dying, and he wanted a solution.
Link was sympathetic, but he also recognized a scientific whodunit waiting to be solved by someone with his expertise. In fact, 12 years earlier in Canada, Frank Schofield, a veterinary pathologist working at the Ontario Veterinary College had concluded that the strange hemorrhaging illness correlated with wet summers, when unusually damp hay used as cattle feed became infected with fungal molds. More specifically, he connected the disease to silage containing large amounts of moldy sweet clover. The Penicillium molds on their own were harmless. Instead, Schofield proposed that the interaction of sweet clover with the infecting fungal strains created a potent anticoagulant in the cows’ blood, causing them to bleed out. But no one was able to isolate and identify the hypothetical substance causing “sweet clover disease,” which continued to decimate cattle herds.
Schofield recommended a seemingly easy remedy: farmers simply had to substitute fresh hay for moldy hay in their cows’ silage. By the 1930s some farmers had successfully followed his advice, but for many, such as Ed Carlson, disposing of feed that was only slightly spoiled presented an economic hardship they could ill afford in the lean times marking the beginning of the Great Depression. And many were skeptical of Schofield’s theories about moldy sweet clover because they had never had a problem with their pastures or silage before.

Looking at the dead and still bloody cow on his laboratory floor, Link remembered Schofield’s publications on poisoning by sweet clover. Already a renowned expert in plant carbohydrate chemistry, Link decided to see for himself if there was a mysterious hemorrhagic agent lurking in the spoiled hay. Over the next six years Link’s students isolated candidate compounds from hay samples and tested them one by one for anticlotting activity in lab animals. Finally, in June 1939, Link’s student Harold Campbell isolated a substance that reduced clotting. Its molecular formula was C19H12O6, also known as 3,3′-methylenebis (4-hydroxycoumarin). They called the compound dicoumarol because it had a structure similar to coumarin, a bitter-tasting substance found in such plants as cinnamon and especially abundant in sweet clover. As others later demonstrated, the Penicillium molds convert coumarin, a relatively harmless chemical, into dicoumarol, an anticoagulant that is deadly in large doses.
Link and his students went on to develop a method for synthesizing dicoumarol and, armed with large quantities of the chemical, observed the effects of various doses in lab animals. As Link examined the results, he realized that dicoumarol (or some variant of it) might be of great use on operating tables and in postsurgical wards, preventing potentially fatal blood clots by keeping blood free flowing and thin.
Analog #42 may not have been the answer to life, the universe, and everything, but it would soon revolutionize pest control.
Beginning in 1940 the Wisconsin Alumni Research Foundation (WARF), which funded Link’s lab, made dicoumarol available for clinical experimentation in humans. Medical researchers at Wisconsin General Hospital, the Mayo Clinic, Montreal General Hospital, and elsewhere conducted the first studies of how dicoumarol worked in human bodies. WARF president George Haight himself benefited from dicoumarol following gallbladder surgery. The need for such a drug became even more urgent as World War II hit American shores in 1941. That same year an editorial in the Lancet catapulted dicoumarol onto the international stage. The writers suggested the drug, which could be taken orally, might supersede the primary anticoagulant then in use, heparin, which had to be injected.
Still, dicoumarol was far from a perfect drug. High doses of the “cow poison” were needed to reduce clotting in humans, but too much caused dangerous hemorrhaging. And it was unreliable in emergencies because of its slow absorption into the bloodstream. But Link, whose lab had synthesized more than 100 chemical variants of dicoumarol, had a hunch a better drug was hiding among them.
Some of the analogs Link’s team synthesized proved even more potent than dicoumarol at provoking internal bleeding in the lab’s rats, mice, and rabbits. Researchers for the most part couldn’t visually inspect animals for bleeding, although blood would show up in stool and urine. More often the animals’ external signs of internal bleeding were pain, dizziness or disorientation, and faintness as organ systems shut down; with repeated doses the animals would eventually collapse. One version of the drug, created by Link’s students Miyoshi Ikawa and Mark Stahmann and named “analog #42,” was so much more toxic in lab rats that the researchers began to consider its rodent-killing potential. Link later called this chemical approach to pest control a “better mousetrap.”

WARF helped Link’s lab file a patent for analog #42; Link, in turn, named the compound warfarin, in honor of the foundation. Analog #42 may not have been the answer to life, the universe, and everything, but it would soon revolutionize pest control.
In the summer of 1950 news of Link’s recently discovered rat poison reached 31-year-old Chicago industrialist Lee Leonard Ratner. Recognizing a lucrative business opportunity, Ratner secured a licensing agreement with WARF and formed a company to market warfarin under the brand name d-CON.
Warfarin was much more effective than competing products because it was slow acting but lethal over repeated doses. Rats encountering warfarin did not become bait shy after the first nibble as they did with other, more potent rat poisons, such as strychnine and zinc phosphide. Instead, they kept returning to the bait until they had ingested a toxic dose. At the same time, small quantities had little effect in larger mammals, and regulators deemed it safer to use in the presence of curious children and pets.
In November 1950 Ratner staged a public demonstration in Middleton, Wisconsin. Within two weeks his strategically placed d-CON bait stations had eradicated the town’s chronic rat infestation. Propelled by Ratner’s savvy coast-to-coast radio advertising campaign, d-CON grew into a multimillion-dollar mail-order business. Warfarin, the “Pied Piper” of pest control, became a fixture in homes, church basements, grocery and hardware stores, and the barns and farmsteads of rural America.
In the meantime Link, well aware of dicoumarol’s therapeutic shortcomings, reexamined his data on warfarin and speculated it might prove the better drug. In 1950 he encouraged hematologist colleagues to start clinical trials with warfarin, but they refused, reluctant to test it on humans because of its growing reputation as a proficient and deadly rat killer.

Then in 1951, navy recruit EJH’s unwitting medical experiment changed doctors’ minds about warfarin and effectively ended dicoumarol’s clinical future. Doctors saved EJH from bleeding out by giving him blood transfusions and by treating him with vitamin K, a known antidote to excess dicoumarol. His speedy recovery demonstrated that vitamin K also acted as an antidote to dicoumarol’s chemical cousin warfarin. The availability of an antidote emboldened doctors: warfarin doses sufficient to kill rats might be safe and even therapeutic in patients with clotting disorders. But before doctors could test warfarin on humans, they needed a purer form of the chemical. Link convinced a friend at Endo Laboratories, Samuel Gordon, to manufacture enough clinical-grade warfarin to begin human testing.
Early trials revealed that warfarin could do everything dicoumarol could but with several times the potency. When an arterial blood clot in or near the brain caused a stroke, warfarin was able to restore blood flow and prevent another stroke. And when patients lay in bed for long periods after surgery, warfarin prevented the slow-moving blood in their leg veins from clotting. Whereas dicoumarol’s effects were often delayed and short lived, warfarin had immediate and long-lasting effects, regardless of whether it was delivered orally or intravenously. Paradoxically, the properties that differentiated warfarin from dicoumarol made it both a better rat poison and a better medicine in humans.
Finding the appropriate warfarin dose sometimes feels like a game of Russian roulette—the longer it takes, the higher the chance of deadly complications.
In 1954, three years after EJH’s near-fatal poisoning, the FDA assessed the results of these trials and approved warfarin for the prevention of blood clots leading to strokes in humans. Endo Laboratories, which now owned the patent to warfarin sodium, brought it to market under the brand name Coumadin that same year.
President Dwight D. Eisenhower was the first high-profile patient to be treated with the new anticoagulant. While on vacation at his in-laws’ house in Denver in 1955, Eisenhower suffered a heart attack, which damaged his already weakened heart. His doctors administered Coumadin to counteract clots that might lead to a stroke and kept him on a maintenance dose. Warfarin saved Eisenhower from the possibly fatal aftermath of his heart attack. The media hailed the drug as a miracle blood thinner, and its use in clinics and hospitals rose dramatically.
Today warfarin is one of the most highly prescribed drugs in the United States, an essential item in hospital supply cupboards and on pharmacy shelves. For six decades it has been used to prevent fatal embolisms in patients suffering from hereditary or acquired clotting disorders, survivors of strokes and heart attacks, and postoperative patients recovering from major surgery.
Even as it was hailed as a medical breakthrough, though, warfarin earned a reputation as a tricky drug to prescribe. Some patients needed low doses, around 5 milligrams per week, to control their rates of blood clotting; others needed higher doses, as much as 70 milligrams per week, to achieve the same effect. There was no easy way to predict which end of the spectrum a new patient would be on and no clear biological explanation for why such a wide range of doses was necessary. Researchers speculated that some bodies metabolized the drug quickly, thus needing more, while others metabolized it so slowly that it persisted in the body for longer than average, requiring a correspondingly lower dose.

U.S. Army medics treating soldiers in England before the invasion of Normandy, 1944. Dicoumarol’s anticoagulant effects were badly needed in the battlefield hospitals of World War II.
Such unpredictability aggravated doctors because it had serious consequences. The therapeutic window for warfarin was exceedingly narrow and unique to each individual. Even slight over- or underdosing could quickly lead to complications, such as internal bleeding or strokes. Warfarin consistently ranked among the top drugs whose adverse effects led to hospitalization, especially in older adults, second only to insulin. Major bleeding events occurred in an estimated 10% to 20% of users; up to 1% of patients died each year from complications.
Though there was no magic formula for determining the precise warfarin dose at which each patient’s blood is neither too thin nor too thick, over the years researchers unearthed some clues. In the early 1970s doctors at Albert Einstein College of Medicine of Yeshiva University and the Bronx Municipal Hospital Center, New York, found that age, sex, body size, and other attributes played a role in how the body metabolizes warfarin. Still, warfarin prescribing practices continue to differ widely. Today, many doctors start patients on a standard dose and adjust up or down based on their responses; some make an educated guess at an initial dose based on the patient’s attributes and adjust accordingly. Finding the appropriate warfarin dose sometimes feels like a game of Russian roulette—the longer it takes, the higher the chance of deadly complications.
In 1992 biochemical pharmacologist Richard Okita had his first encounter with warfarin after his father suffered a mini-stroke at Thanksgiving dinner. The Okitas rushed him to the local hospital in Thousand Oaks, California, where, after being stabilized, he was prescribed warfarin. It took doctors three weeks to zero in on the right dose, though fortunately without complications.
As a scientist Okita was investigating the biochemical activities of cytochrome P450 enzymes, which metabolize foreign compounds, including toxins and drugs, in the liver. Though he was studying rats, the enzymes are also found in humans. The same year Okita’s father had his stroke researchers identified a cytochrome P450 enzyme called CYP2C9 that breaks down and neutralizes warfarin.

As early as the 1950s, researchers theorized that the speed with which certain enzymes break down drugs might be modulated by an individual’s DNA. Geneticist Arno Motulsky and pharmacologist Werner Kalow proposed that each person’s unique genetic code specified slightly different versions of proteins and enzymes and that some of these differences led to stronger or weaker responses to certain drugs. In one experiment Kalow showed how the same dose of succinylcholine, a drug that paralyzes muscles, had different effects in different people depending on their particular form of an enzyme called cholinesterase. The work of Kalow and Motulsky laid the foundations of “pharmacogenetics,” the search for genetic explanations for people’s different reactions to medications.
For much of the 20th century their theories saw little practical application in medicine, as human genes remained largely unknown and inaccessible. Not until 1990 and the launch of the Human Genome Project could researchers begin to examine the role of specific genes in warfarin therapy. Biology’s “moonshot” effort to sequence the human genome and identify all its genes meant researchers could finally systematically answer a 40-year-old question: how did genes affect drug responses? And, more important, could genetic information lead to safer, more accurate dosing of life-saving medications?
The body’s remarkable ability to generate within seconds the fine matrix of fibers that constitute a blood clot is a delicate, complicated process. It’s known as a molecular cascade, a sequence of almost 30 enzymes and proteins that act one after the other to change blood from a liquid to a gel, forming a plug that stops blood loss at the site of injury. Perturb even one of these steps and the whole cascade stops in its tracks.
In the 1990s researchers finally identified what Link, who died in 1978, would surely have found exciting—the gene for warfarin’s molecular target in the body. Warfarin interferes with the proper functioning of a blood-clotting enzyme called VKORC1, preventing the accumulation of the vitamin K needed to create clots and to stem bleeding. In pharmacological parlance warfarin is a VKORC1 inhibitor. Researchers reasoned that if some patients’ DNA encoded a slightly altered version of the VKORC1 enzyme that was less affected by warfarin, then they would need a higher dose. In contrast, a DNA alteration that rendered the enzyme more susceptible would render warfarin more potent in those patients, requiring a lower dose. Similarly, researchers predicted that individuals whose DNA specified less active versions of CYP2C9, the enzyme that metabolizes warfarin, would need a lower dose.
Warfarin remains one of the most highly prescribed drugs in the United States, with tens of millions of prescriptions written and two million new patients put on it annually. As the nation’s demographics shift to older populations, this number is only expected to go up since the elderly are more prone to the heart attacks, strokes, and surgeries for which warfarin is most commonly prescribed. If DNA sequence information could improve warfarin dosing, the impact on health care in the United States could be enormous, potentially saving millions of dollars and many lives.
This sense of urgency catalyzed new programs at the National Institute of General Medical Sciences in the early 2000s, including new programs at the Division of Pharmacology, Physiology, and Biological Chemistry, of which Okita is now a program director. Rochelle Long, director of the division, says, “The whole point of these efforts was that, through the use of pharmacogenetics, you could predict who was going to have an adverse reaction in advance, before they had it.” Warfarin dosing became an important test case for assessing the value of genetic information in making initial treatment decisions.

Mayo Clinic technician Paul Heimgartner performs computer analyses of “genetic signatures” to predict how individuals metabolize and respond to warfarin, which in turn help doctors find the correct dosage for the individual, 2006.
Between 2000 and 2008 researchers studied thousands of human DNA samples and identified genetic sequence variants in the VKORC1 and CYP2C9 genes. As predicted, many of these variants translate to functionally unique versions of the enzymes, with different capacities to interact with warfarin. This molecular picture explained in part why warfarin had different effects in different bodies. To test the clinical value of this genetic information, clinics around the United States and from eight other countries combined their data and DNA samples to create the International Warfarin Pharmacogenomics Consortium (IWPC). In 2009 the IWPC examined data from thousands of patients receiving warfarin therapy and concluded that genetic testing could make warfarin dosing more accurate. The greatest benefits were seen by patients “at the extremes,” those individuals in the population whose bodies metabolize medications exceedingly quickly or slowly.
The next year, the FDA approved an updated label for warfarin, recommending that clinicians perform DNA testing for all patients new to the drug. Warfarin became the first medication in American history to explicitly carry an FDA recommendation for genetic testing before dosing. The drug became the epicenter of the precision medicine movement, with its promise of healthier lives through detailed knowledge of DNA. Advocates in the biotech and pharmaceutical industries, as well as some researchers and health-care administrators, began imagining an era when patients could visit a pharmacy and be prescribed a cocktail of medications tailored to their genome. Genetic-testing companies began to develop commercial DNA tests for warfarin-related genes that clinics could order for their patients.
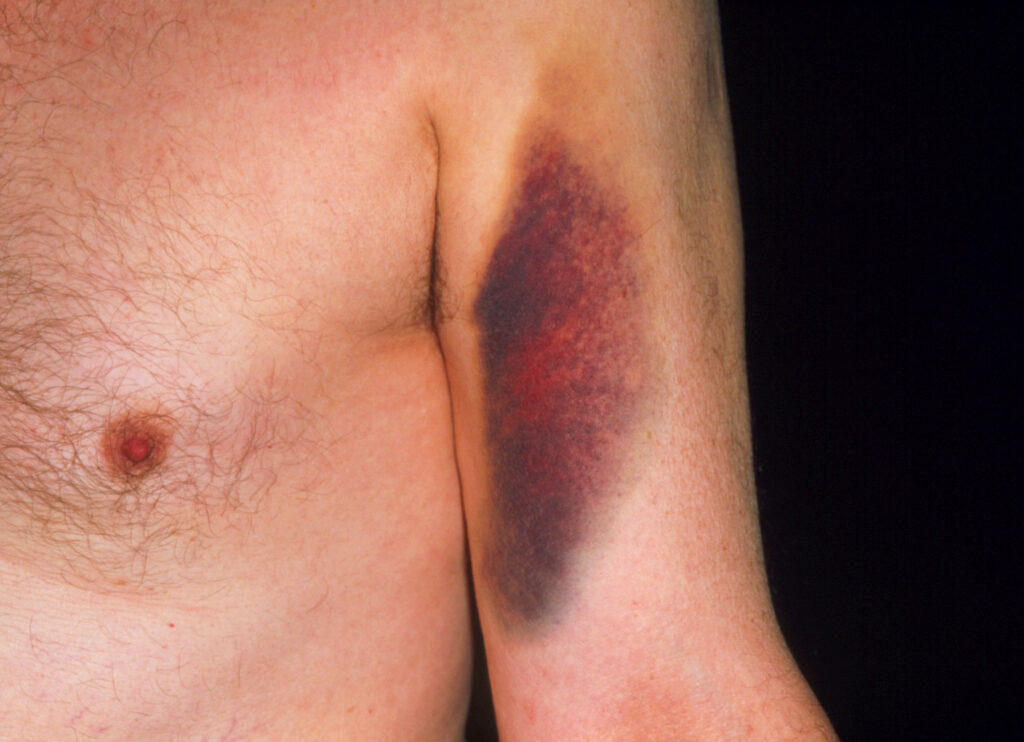
Bruising caused by warfarin. Among older adults complications from warfarin use cause the most drug-related visits to the emergency department.
Despite these hopes few clinics bought into genetic tests for warfarin dosing. The cost of DNA testing is significantly less today than it was just a few years ago, or even last year, but it remains expensive. Insurers are wary of reimbursing the hundreds of dollars each test costs, given the lack of concrete numbers on how many lives or dollars can be saved. To make matters worse two landmark clinical trials reported in December 2013 that DNA information did not significantly improve anticoagulant dosing decisions nor significantly reduce the number of bleeding complications. The trials were run using a small number of patients at well-resourced hospitals with skilled clinicians. At such hospitals much of the work of monitoring patients for side effects and tweaking dosage falls to teams of blood specialists in dedicated anticoagulation clinics. Observers at the FDA reasoned that in such settings local professional expertise may have diminished any additional benefit provided by DNA information. They suggested repeating the trials at rural hospitals where equipment, staff, and resources were more limited and where DNA information could compensate for these limitations and perhaps improve care while decreasing costs.
Okita oversees several National Institutes of Health grants currently supporting such studies of warfarin dosing in small and rural communities. “My father was lucky because he lived two blocks from the hospital where he was going to have his clotting levels tested twice a week to set up his initial dose,” says Okita. “But if you’re living in a rural area, then you may be miles from the hospital setting. Then what happens?” For those people, getting the warfarin dose right the first time really is a matter of life and death, and genetic information has the potential to save lives if the costs can be minimized.
Advocates in the biotech and pharmaceutical industries, as well as some researchers and health-care administrators, began imagining an era when patients could visit a pharmacy and be prescribed a cocktail of medications tailored to their genome.
Genetic research on warfarin and other drugs has helped uncover perhaps the biggest lesson of the genomic era: precise knowledge of our DNA sequences may not be the hoped-for magic bullet. While genetic information can be valuable for some people and in some clinical situations, it is but one piece of the puzzle. Variability in warfarin dose requirements is partly due to DNA and partly due to differing environmental and social conditions. Diet, for example, can affect required dosage. The hospital nutritionist told Okita’s father he had to restrict consumption of dark green vegetables and green tea, both staples in his diet, because their high vitamin K content neutralizes the therapeutic effect of warfarin. Maintaining a stable diet on warfarin is essential; travel or weight-loss programs, if they lead to a change in eating habits, can affect a person’s warfarin sensitivity and their ideal dose.

A nurse steadies a patient during physical therapy, undated. Despite scary side effects and difficulties in dosing, warfarin’s ability to prevent strokes and heart attacks is why it remains one of the most prescribed drugs in the United States.
If it holds true that genetic information is most useful for patients at the extremes of warfarin dosing ranges, then the era of pharmacogenomics presents a catch-22; some individuals may see life-saving benefits from genetic testing before their initial dose—but we won’t know who they are until they get tested. For the rest, as Long says, “Even knowing that one is not an ‘exceptional’ person at risk is useful clinical information.” So the value for the average patient will depend on how quickly the costs of genetic testing become universally affordable.
Understanding the relationships between genes and other prescription medications is an active area of research. For example, DNA information can improve clinical decisions about which cancer drugs will work for certain types of cancer, such as lymphomas. As Long points out, clinical pharmacogenomics may be more cost-effective in the long run if patients are tested simultaneously for many genes affecting many drugs. She adds that the “costs of sequencing an entire genome are dropping rapidly, so in the future, it is plausible that the entire DNA sequence will be determined at birth and entered into the health record.”
DNA-based warfarin therapy has been adopted at a few hospitals and by some health providers. Meanwhile, research on the cost-effectiveness of DNA-guided dosing is ongoing. Warfarin itself continues to play an essential role in modern medicine. Newer, competing drugs have tried to displace it, with promises of greater safety, but with larger price tags and spottier insurance coverage. The new drugs work better for some patients, but these medications come with their own side effects. They also lack the six decades of expertise, data, and research that accompanies warfarin. Whatever its complications warfarin remains the gold standard of anticoagulants.